The secret life of single-atom catalysts

Precious metals represent efficient catalysts for a range of technologically and energetically relevant chemical reactions. Catalytic reactions only take part on the surface of a metal. Therefore, catalysts usually take the form of metal nanoparticles to maximize the surface area exposed to reactants. Currently we observe a rapid development of so-called single-atom catalysts where precious metals are dispersed to single isolated atoms so that the catalytic action can be potentially provided by each precious metal atom.
This is the end of the easy part of the story. Single-atom dispersion is energetically unfavorable and a suitable substrate is required to provide strong enough bonding to metal single atoms to preserve their separation. Since we deal with single atoms, the strength of the bond is defined by the local morphology and chemical nature of the substrate on the atomic scale. Single-atom catalysts thus represent highly dynamic and difficult-to-grasp systems readily changing their structure from metal single atoms to nanoparticles and vice versa upon exposure to different reactants, between preparation and utilization, or even between utilization and characterization.
In the highlighted study, researchers investigated the stability of Pt nanoparticles supported on ceria in the dynamically changing oxidizing and reducing reaction atmospheres (Figure 1). Atomic-level insight has been obtained using single-crystalline flat CeO2(111) surface and experimental methods of surface science including scanning tunneling microscopy (STM), together with density functional theory-based calculations (Figure 2).

Figure 1: Reversible changes of Pt morphology on CeO2(111) single-atom catalyst substrate from Pt nanoparticles at reducing conditions (a, c) to Pt single-atoms at monoatomic step-edges at oxidizing conditions (b, d). a, b: STM micrographs, 30 x 30 nm2, c, d: KMC simulated morphologies. Adapted with permission from M. Farnesi Camellone et al., ACS Catal. 12, 4859 (2022). Copyright 2022 American Chemical Society.
“Density-functional theory calculations performed by Matteo Farnesi Camellone and Stefano Fabris at NFFA were crucial for understanding how Pt single atoms interact with single-atom traps at the catalyst substrate.”
“Density-functional theory calculations performed by Matteo Farnesi Camellone and Stefano Fabris at NFFA were crucial for understanding how Pt single atoms interact with single-atom traps at the catalyst substrate and how this interaction, and, consequently, the structure of the catalyst, depends on the reaction atmosphere.”
The research resulted in formulating a generic model of Pt coarsening and redispersion on CeO2(111) surface. Kinetic Monte-Carlo (KMC) implementation of the model successfully mimics the dynamic phenomena of Pt redispersion and coarsening (Figs. 1c, d) and reveals that any state of the Pt deposit on ceria is a result of the competition for Pt atoms between metallic clusters and single atom traps.
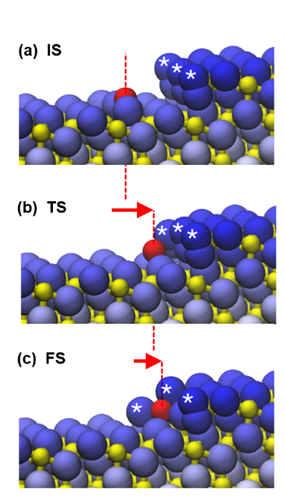
DFT simulation of the capture of Pt single-atom at CeO2(111) monoatomic step-edge. (a) initial state, (b) transient state, (c) final state. Asterisks mark the superoxo species present at oxidizing conditions and bringing the essential stability to the captured Pt atom. Adapted with permission from M. Farnesi Camellone et al., ACS Catal. 12, 4859 (2022). Copyright 2022 American Chemical Society.
"In a sense, our study is removing the boundary between single-atom and traditional supported metal catalysts asking “How would you like to have it?” and showing ways to dynamic control of on-demand catalyst structure.”
“The results of our study allow us to envision regenerating the morphology of Pt/ceria catalysts deactivated in a harsh catalytic process or even sinter-resistant catalysts working in alternating oxidizing/reducing conditions. In a sense, our study is removing the boundary between single-atom and traditional supported metal catalysts asking “How would you like to have it?” and showing ways to dynamic control of on-demand catalyst structure.”
Matteo Farnesi Camellone, Filip Dvořák, Mykhailo Vorokhta, Andrii Tovt, Ivan Khalakhan, Viktor Johánek, Tomáš Skála, Iva Matolínová, Stefano Fabris,and Josef Mysliveček,
ACS Catal. 2022, 12, 4859−4871, https://doi.org/10.1021/acscatal.2c00291
Theory and Simulation Installation 6
STRUCTURAL AND GROUND-STATE ELECTRONIC PROPERTIES
ATOMS AND MOLECULES IN MOTION
at CNR-IOM
Josef Mysliveček
Josef Myslivečekis Associated Professor of surface science at Charles University in Prague, Czech Republic. His expertise is centered around bottom-up nanopatterning of metal, semiconductor, and oxide surfaces. The resulting nanostructures find applications in the fields of electronics, molecular electronics, heterogeneous catalysis, and electrocatalysis. In recent years, he has developed metal-oxide model catalytic systems with adjustable density of active sites, oxygen vacancies, monoatomic steps, and supported single atoms. Morphology, chemical composition, chemical state, and crystallographic structure of the samples are determined with atomic resolution by experimental surface science techniques. The obtained quantitative morphological information is essential for the advanced studies of the structure-property relationships in heterogeneous catalysis and in electrocatalysis. Josef Mysliveček has authored or co-authored >60 publications with >2000 citations and his h-index is 22.

